Table Of Content

In order to translate these techniques to clinical practice, the next step would be to conduct a pivotal trial. In this review we discuss the underlying principles of clinical trial design with an emphasis on assessing design risks that lead to trial failure as well as negative trials. While of general interest, this is perhaps particularly timely for the neuromodulation community, given the paucity of well-designed trials in the field. Clinical trials are a fundamental component of medical research and serve as the main route to obtain evidence of the safety and efficacy of treatment before its approval. A trial's ability to provide the intended evidence hinges on appropriate design, background knowledge, trial rationale to sample size, and interim monitoring rules.
Challenges and Opportunities for Designing Clinical Trials for Antibody Mediated Rejection
A well-designed and executed clinical trial is the gold standard of evidence-based medicine. It is important for readers to understand the rationale for the study design, identify common pitfalls, and scrutinize limitations. Herein, we present a brief overview of types of designs used for clinical trials and discuss the use of appropriate end points, the selection of study participants, randomization, sample size calculation, blinding, and analysis of data. Our goal is to provide a primer for practicing urologists to enhance their understanding of the clinical trial literature. Clinical trials are experiments designed to evaluate new interventions to prevent or treat disease in humans.
Unique Trial Design Expected to be Focus of Upcoming Donanemab Adcomm - BioSpace
Unique Trial Design Expected to be Focus of Upcoming Donanemab Adcomm.
Posted: Tue, 12 Mar 2024 07:00:00 GMT [source]
Design and Interpretation of Clinical Trials
We can advise you on the best group options to meet your organization’s training and development goals and provide you with the support needed to streamline the process. Participating together, your group will develop a shared knowledge, language, and mindset to tackle the challenges ahead. Unplanned unblinding should only be undertaken to protect participant safety (i.e., if the treatment assignment is critical for making immediate therapeutic decisions). 5 patients aged between 6 and 10 have been treated to date with GNT0004, 4 in France and 1 in the UK.
Professional development
All authours accept responsibly for the content of the final typescript and agree to submit for publication. All rights are reserved, including those for text and data mining, AI training, and similar technologies. Researchers should explicitly state whether a study was blinded, who was blinded, how blinding was achieved, the reasons for any unplanned unblinding, and state the results of an evaluation of the success of the blinding.
Medical
Historical controls are obtained from studies that have already been conducted and are often published in the medical literature. The data for such controls is external to the trial being designed and will be compared with data collected in the trial being designed. The advantage of using historical controls is that the current trial will require fewer participants and thus use of historical controls provides an attractive option from a cost and efficiency perspective. Historical controls are rarely used in clinical trials for drug development due to the concerns for bias.
Features such as the open-label design and the comparative effectiveness approach that forgoes placebo permit flexibility, particularly with variable routes of administration (intravenous, subcutaneous, inhaled). Moreover, we determined at the outset to seek strong signals of efficacy, while accepting the risk of missing more modest benefits in exchange for the goal of rapidly cycling and testing several agents at a time. These design decisions have enabled fast progress, with the caveat that I-SPY COVID is signal-finding rather than definitive and so subsequent phase 3 studies will be required for agents that graduate from the trial. Similar approaches may be useful in future pandemic settings when disease mechanisms are poorly defined, multiple agents need to be rapidly triaged, and/or there is potential for major therapeutic wins.
Avoiding designing failed clinical trials: main issues that lead to invalid data
Advances in statistical software and computing power continue to allow for increasingly more complex study designs and analytical techniques, and researchers should take best advantage of these advances. There is a concern regarding acceptability of evidence generated by alternative trial designs by regulatory authorities and peer reviewers. It is imperative to understand that same research question may be tackled through alternative designs and that there is no definitive trial design for every research question. The time frame, logistics involved, and availability of study subjects are key to selection. Factors, such as objective of the trial, number of patients needed, length of trial, and how the variability is handled, could be important in the choice of the most suitable trial design.
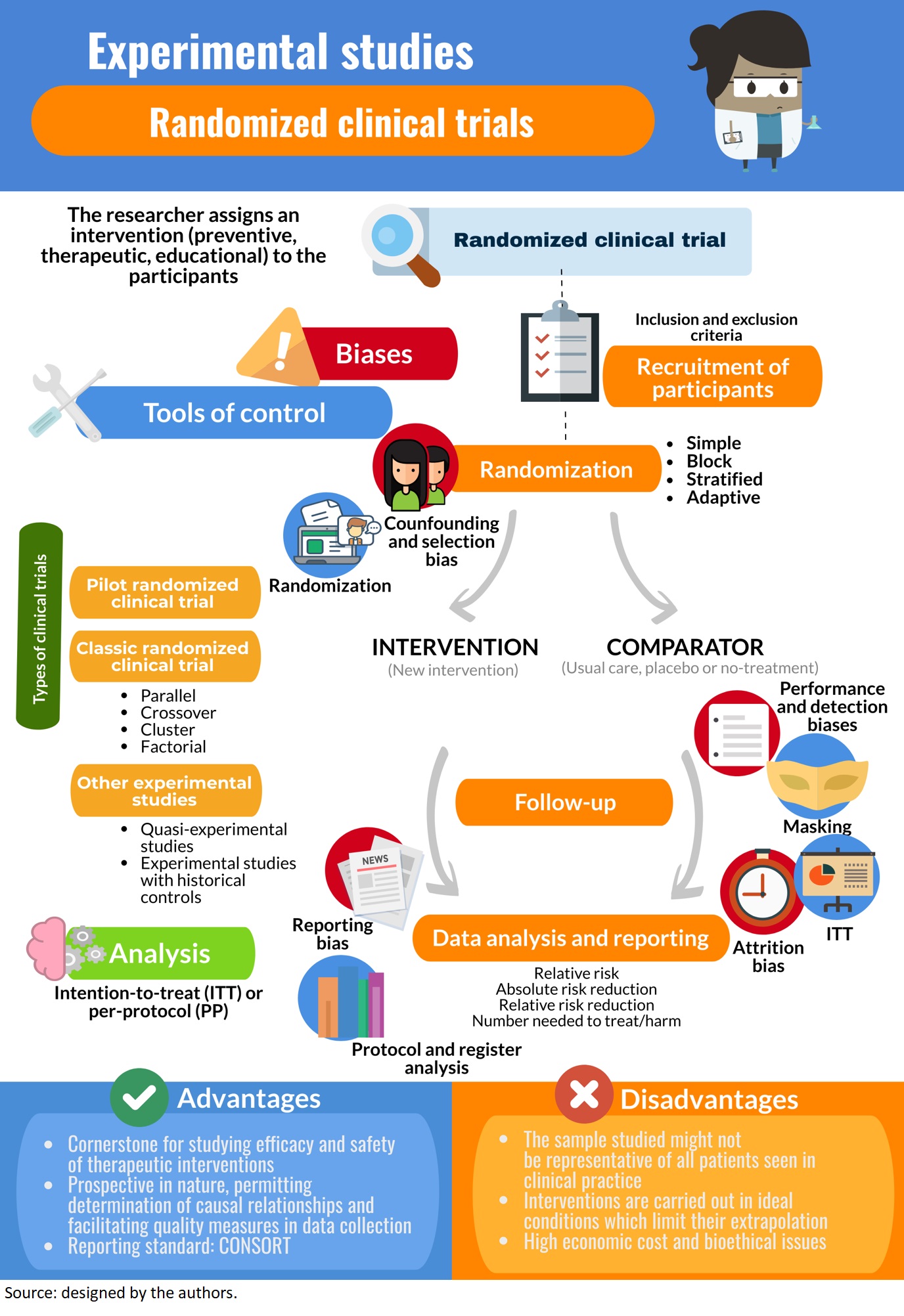
One of the aspects that is often overlooked is the selection of cases and controls. It is important to select the cases and controls appropriately to obtain a meaningful and scientifically sound conclusion and this can be achieved by implementing matching. Even upon following best practices in trial design, the investigator needs to be prepared for unexpected roadblocks such as lack of funding, challenges in training personnel, recruitment, and other factors diminishing trial feasibility.
Overview of clinical trial phases
(ii) N of 1 design – N of 1 trials or “single-subject” or “structured within-patient randomized controlled multi-crossover trial design” are used to evaluate all interventions in a single patient. A typical single patient trial consists of experimental/control treatment periods repeated a number of times. Usually, the primary objective of such a trial is to determine the treatment preference for the individual patient and this design is gaining popularity in recent times.
Has a grant from Merck for an Investigator-initiated trial of ductal carcinoma in situ. Received funding for this article from QHLC, as well as other funding from the NIH and Roche/Genentech. A.D..B is a member of the Scientific Advisory Board Committee of Caris Life Sciences. Thus, while designing a study it is important to take measure to limit bias as much as possible so that the scientific validity of the study results is preserved to its maximum. Hence, while designing a research study, both the scientific validity and ethical aspects of the study will need to be thoroughly evaluated. Following the positive opinion of the DMC (Data Monitoring Committee), Genethon is preparing the pivotal European phase of the trial with the European Medicines Agency (EMA).
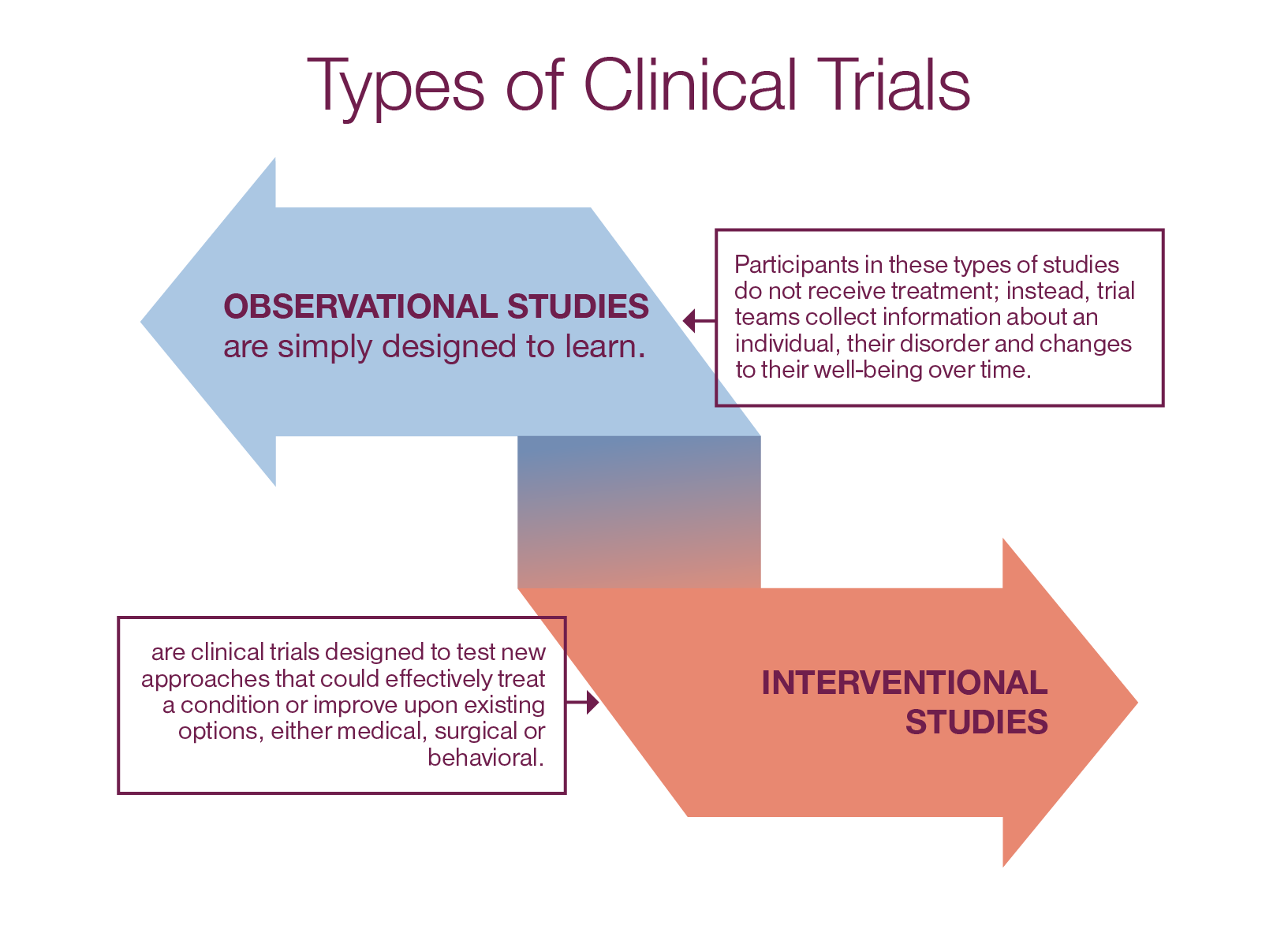
It involves an intervention that tests the association between the exposure and outcome. Each study design is different, and so it would be important to choose a design that would most appropriately answer the question in mind and provide the most valuable information. A study’s risk-benefit ratio should always include safety outcomes, whether they are clinical, surrogate or both. Patient safety must be prioritized over trial completion, and a well-designed clinical trial is less likely to be terminated early for safety reasons.
Single-arm designs – wherein all patients receive the same intervention and are generally compared to a historical control group – can provide some information on treatment effect. However, often the single arm of patients and the historical control group do not represent the same populations of interest nor receive treatment under similar trial conditions. As such, single-arm designs are limited in the conclusions they can draw and less desirable than randomized trials. In randomized trials, there are at least 2 treatment groups (or “arms”) to which patients are randomly assigned. The random assignment, or randomization, aims to create groups that are similar with respect to all factors, besides the intervention, that might affect the outcome. Common examples include the use of a control arm (i.e. an arm that receives the standard of care) and blinding (i.e. patient and/or clinician do not know the treatment assignment) to reduce bias.
Any interpretation of a non-inferiority trial requires the reader to evaluate whether they find the non-inferiority margin selected convincing and of clinical import. It is critical to assess how the results of a trial will better the field if its aims are achieved – before selecting these aims. An investigator must carefully consider the problem the study is trying to solve. The more prevalent and devastating the problem is, and the greater the potential benefits, the easier it is to justify performing the trial.
For example a trial might be designed to estimate the difference in response rates between two therapies with appropriate precision. Appropriate precision might be measured by the width of a confidence interval of the difference between the two response rates. The Company will host a conference call today where HOOKIPA’s Executive Team will discuss the full details of the trial design, and the Company’s clinical development strategy for oncology. Once the design elements in the conceptual phase have been identified, and there is consensus to move forward with designing a clinical trial, the design elements necessary for actually running the trial need to be specified. This constitutes the implementation phase, the steps for which are outlined below. A trial’s ability to answer its research question is enhanced by having its hypothesis built on coherent preliminary data.
Adherence to the trial protocol also has a significant impact on internal validity, and measures should be taken to maintain adherence, e.g. checklists, and careful selection of exclusion criteria. Although the phase design framework helps the investigator to choose the appropriate phase and thus design the study based on its main objectives, the choice of phase is not always easy to determine. For example, the amount of phase II data needed in order to move to a phase III trial is not always clear.
Sample size considerations for Bayesian designs depend on additional factors, most notably the prior distribution of the effect size; see Pezeshk (2003). Finally, our experience in I-SPY COVID has taught us that it is possible to balance pragmatism, safety and discovery in the context of a phase 2 trial, even during a pandemic. I-SPY COVID takes a moderately pragmatic approach to streamlined data collection, with a standardized method to ascertain adverse events and outcomes across all arms. An observational cohort of patients who meet trial criteria but are not randomized provides a real-world comparator arm.







No comments:
Post a Comment